Nel
precedente post abbiamo visto come si può costruire una base curvilinea locale a partire da una base cartesiana e viceversa, in particolare
abbiamo ottenuto le seguenti relazioni tra le basi dei due riferimenti:
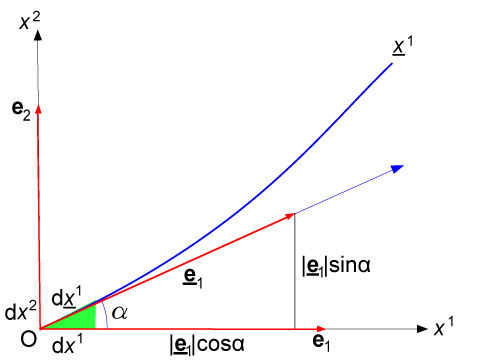
ei=(∂xj/∂xi)ej , ei=(∂xj/∂xi)ej
dove ei sono le basi del riferimento cartesiano (x1,..., xn) mentre ej sono le basi del riferimento curvilineo (x1,..., xn) in uno spazio Rn (con i,j=1,..., n).
In questo caso particolare i valori dei termini ∂ei/∂xj espressi in funzione di er e eθ sono (con i,j=r,θ):
Si osservi in particolare che se supponiamo che le componenti di T lungo r e θ non variano (cioè se ∂Ti/∂r=0 e ∂Ti/∂θ=0) si ottiene:
[Una ottima esposizione di questi concetti si trova nella Playlist Video di Dermot Green - Queen's University Belfast]
Nota:
come sempre le coordinate curvilinee sono definite in funzione di quelle
cartesiane e viceversa, inoltre sono funzioni differenziabili.
È interessante osservare che possiamo riscrivere le relazioni sopra (raccogliendo componenti x basi) come:
È interessante osservare che possiamo riscrivere le relazioni sopra (raccogliendo componenti x basi) come:
ei=(∂xjej)/∂xi=∂r/∂xi , ei=(∂xjej)/∂xi=∂r/∂xi
dove si è posto ∂r=∂xjej=∂xjej e quindi il differenziale dr del raggio vettore r si può scrivere, nei due riferimenti, come basi x componenti:dr=(∂r/∂xj)dxj =ejdxj oppure dr=(∂r/∂xj)dxj=ejdxj.
Nota: dr è un vettore e quindi deve restare invariato nei due riferimenti.
Ricordiamo inoltre che in generale un vettore T si può definire in funzione delle sue basi ei e delle sue componenti Ti:
Ricordiamo inoltre che in generale un vettore T si può definire in funzione delle sue basi ei e delle sue componenti Ti:
T=T1e1+...+Tnen=Tiei
dove abbiamo applicato la notazione di Einstein sugli indici i=1,..., n.
Abbiamo ricordato sopra che se cambiamo riferimento le basi cambiano come ej=(∂xi/∂xj)ei quindi se vogliamo che il vettore T=Tjej resti invariato anche le sue componenti Tj dovranno trasformarsi (in modo inverso):
Abbiamo ricordato sopra che se cambiamo riferimento le basi cambiano come ej=(∂xi/∂xj)ei quindi se vogliamo che il vettore T=Tjej resti invariato anche le sue componenti Tj dovranno trasformarsi (in modo inverso):
Tj=(∂xj/∂xi)Ti
in modo cioè che nel nuovo riferimento il vettore T resti invariato:
T=Tjej=(∂xj/∂xi)Ti(∂xi/∂xj)ei=Tiei=T
essendo (∂xj/∂xi)(∂xi/∂xj)=1.
Nota: dal differenziale dxj=(∂xj/∂xi)dxi segue dxj/dxj=(∂xj/∂xi)(∂xi/∂xj)=1.
Calcoliamo ora la derivata del vettore T=Tiei lungo una coordinata xj qualsiasi, questa sarà definita (come derivata di una funzione prodotto):
Calcoliamo ora la derivata del vettore T=Tiei lungo una coordinata xj qualsiasi, questa sarà definita (come derivata di una funzione prodotto):
∂T/∂xj=∂(Tiei )/∂xj=(∂Ti/∂xj)ei+Ti(∂ei/∂xj)
dove il termine ∂ei/∂xj tiene conto della possibile variazione delle basi ei rispetto alle coordinate xj: questa derivata è detta derivata covariante e in pratica estende il concetto usuale di derivata direzionale.
Nota: si dice derivata covariante perché preserva il carattere di invarianza rispetto alla trasformazione di coordinate (vedi il relativo post).
Si osservi che se le coordinate sono cartesiane, allora le basi non variano in funzione delle coordinate (cioè mantengono sempre stesso modulo e direzione in ogni punto e quindi ∂ei/∂xj=0); in tal caso la derivata si riduce alla classica derivata direzionale (calcolata cioè lungo l'asse coordinato xj):
Nota: si dice derivata covariante perché preserva il carattere di invarianza rispetto alla trasformazione di coordinate (vedi il relativo post).
Si osservi che se le coordinate sono cartesiane, allora le basi non variano in funzione delle coordinate (cioè mantengono sempre stesso modulo e direzione in ogni punto e quindi ∂ei/∂xj=0); in tal caso la derivata si riduce alla classica derivata direzionale (calcolata cioè lungo l'asse coordinato xj):
∂T/∂xj=(∂Ti/∂xj)ei
dove ricordiamo T è un vettore n-dimensionale (con i,j=1,..., n).
Tuttavia nel caso più generale di coordinate curvilinee il modulo e la direzione delle basi può variare da punto a punto e quindi il termine ∂ei/∂xj è generalmente diverso da zero*.
Vediamo quindi un esempio riprendendo le coordinate curve polari (r,θ) introdotte nel precedente post e le relative basi (er,eθ) ottenute in funzione delle coordinate cartesiane (ex,ey):
Tuttavia nel caso più generale di coordinate curvilinee il modulo e la direzione delle basi può variare da punto a punto e quindi il termine ∂ei/∂xj è generalmente diverso da zero*.
Vediamo quindi un esempio riprendendo le coordinate curve polari (r,θ) introdotte nel precedente post e le relative basi (er,eθ) ottenute in funzione delle coordinate cartesiane (ex,ey):
er=cosθex+sinθey
eθ=-rsinθex+rcosθey.
ricordando che il vettore T si esprime come (con i=r,θ):
T=Tiei=Trer+Tθeθ.
In questo caso particolare i valori dei termini ∂ei/∂xj espressi in funzione di er e eθ sono (con i,j=r,θ):
∂er/∂r=∂(cosθex+sinθey)/∂r=0
∂er/∂θ=-sinθex+cosθey=(1/r)eθ
∂eθ/∂r=-sinθex+cosθey=(1/r)eθ
∂eθ/∂θ=-rcosθex-rsinθey=-rer
∂er/∂θ=-sinθex+cosθey=(1/r)eθ
∂eθ/∂r=-sinθex+cosθey=(1/r)eθ
∂eθ/∂θ=-rcosθex-rsinθey=-rer
dove notiamo che ∂er/∂θ=∂eθ/∂r e infatti in generale risulta:
∂ei/∂xj=∂ej/∂xi.
Nota: ciò accade in generale quando le derivate seconde incrociate sono uguali**, per le funzioni lisce questa condizione è sempre soddisfatta.
Perciò le derivate rispetto ad r e θ del vettore T sono (con i=r,θ):
Perciò le derivate rispetto ad r e θ del vettore T sono (con i=r,θ):
∂T/∂r=∂(Tiei )/∂r=(∂Ti/∂r)ei+Tr(∂er/∂r)+Tθ(∂eθ/∂r)
∂T/∂θ=∂(Tiei )/∂θ=(∂Ti/∂θ)ei+Tr(∂er/∂θ)+Tθ(∂eθ/∂θ)
∂T/∂θ=∂(Tiei )/∂θ=(∂Ti/∂θ)ei+Tr(∂er/∂θ)+Tθ(∂eθ/∂θ)
e quindi sostituendo i valori delle derivate delle basi ottenuti sopra:
∂T/∂r=(∂Ti/∂r)ei+(1/r)Tθeθ
∂T/∂θ=(∂Ti/∂θ)ei+(1/r)Treθ-rTθer.
∂T/∂θ=(∂Ti/∂θ)ei+(1/r)Treθ-rTθer.
Si osservi in particolare che se supponiamo che le componenti di T lungo r e θ non variano (cioè se ∂Ti/∂r=0 e ∂Ti/∂θ=0) si ottiene:
∂T/∂r=(1/r)Tθeθ
∂T/∂θ=(1/r)Treθ-rTθer
∂T/∂θ=(1/r)Treθ-rTθer
in questo caso si ha cioè il contributo dovuto alla sola variazione delle basi.
Nota: è evidente che il punto r=0 deve essere escluso, infatti qui le coordinate non sono invertibili come richiesto***.
Se infine vogliamo che T venga trasportato parallelamente rispetto alla superficie curva, dovremo annullare la derivata covariante ponendo:
Nota: è evidente che il punto r=0 deve essere escluso, infatti qui le coordinate non sono invertibili come richiesto***.
Se infine vogliamo che T venga trasportato parallelamente rispetto alla superficie curva, dovremo annullare la derivata covariante ponendo:
∂T/∂r=0 e ∂T/∂θ=0
cioè dovremo annullare i valori delle componenti delle derivate parziali ottenuti sopra, rispettivamente lungo er ed eθ.
(*) Solitamente nella derivata covariante per indicare i termini (∂ei/∂xj) si usano i simboli di Christoffel del secondo tipo così definiti: Γkij=(∂ei/∂xj)ek. Perciò ∂T/∂xj=(∂Ti/∂xj)ei+TiΓkijek=(∂Ti/∂xj+TkΓikj)ei (scambiando k con i).
(**) Dalle seguenti relazioni tra basi (vedi il precedente post):
er=(∂x/∂r)ex+(∂y/∂r)ey e eθ=(∂x/∂θ)ex+(∂y/∂θ)ey si ottiene derivando
(**) Dalle seguenti relazioni tra basi (vedi il precedente post):
er=(∂x/∂r)ex+(∂y/∂r)ey e eθ=(∂x/∂θ)ex+(∂y/∂θ)ey si ottiene derivando
∂er/∂θ=(∂x/∂r∂θ)ex+(∂y/∂r∂θ)ey e eθ/∂r=(∂x/∂θ∂r)ex+(∂y/∂θ∂r)ey
da cui si ha: ∂er/∂θ=eθ/∂r se ∂x/∂r∂θ=∂x/∂θ∂r e ∂y/∂r∂θ=∂y/∂θ∂r (cvd).
da cui si ha: ∂er/∂θ=eθ/∂r se ∂x/∂r∂θ=∂x/∂θ∂r e ∂y/∂r∂θ=∂y/∂θ∂r (cvd).
(***) Condizione generale affinché le coordinate siano invertibili è che il determinante della matrice Jacobiana non si annulli.
[Una ottima esposizione di questi concetti si trova nella Playlist Video di Dermot Green - Queen's University Belfast]